|
|
||||
|
Published by : PROFESSIONAL MEDICAL PUBLICATIONS |
||||
|
ISSN 1681-715X |
||||
|
||||
|
- |
||||
|
CASE REPORT |
||||
|
- |
||||
|
Volume 22 |
July - September 2006 |
Number 3 |
||
|
|
||||
|
|
||||
|
|
||||
|
Published by : PROFESSIONAL MEDICAL PUBLICATIONS |
||||
|
ISSN 1681-715X |
||||
|
||||
|
- |
||||
|
CASE REPORT |
||||
|
- |
||||
|
Volume 22 |
July - September 2006 |
Number 3 |
||
|
|
||||
|
|
||||
Anomalous Origin of Left Main Coronary Artery
from Pulmonary Artery (ALCAPA)
Maad Ullah*
ABSTRACT
Anomalous origin of left main coronary artery from pulmonary artery (ALCAPA) is a very rare congenital anomaly, reported in less than 0.5% of all the congenital heart diseases. Left untreated, majority of the patients die in infancy of myocardial ischemia. We report a case with this anomaly, presented in early infancy with progressive dyspnoea, feeding difficulty and cardiomagaly on X-ray. The diagnosis was made on echocardiography and ECG. The baby is surviving on medications, parents having refused the surgical option.
KEY WORDS: Left coronary artery, Pulmonary artery, Myocardial ischemia, Dilated cardiomyopathy.
Pak J Med Sci July - September 2006 Vol. 22 No. 3 313-315
* Dr. Maad Ullah,
Department of Paediatric Cardiology,
Armed Forces Institute of Cardiology &
National Institute of Heart Diseases
(AFIC/NIHD),
Rawalpindi.
Correspondence:
Dr Lt. Col. Maad Ullah,
House# 30, Brick Field Road,
Off. Abid Majeed Road,
Rawalpindi Cantt,
Rawalpindi - Pakistan.
E-mail: maad107@yahoo.co.uk
* Received for Publication: July 25, 2005
* Accepted: February 10, 2006
INTRODUCTION
Anomalous origin of left main coronary artery (LCA) from pulmonary artery is a rare congenital malformation leading to myocardial ischemia in the early infancy. Diagnosis before left ventricular (LV) dysfunction, though important is quite difficult. The younger age and nonspecific symptoms of irritability and feeding problems make it difficult to diagnose even after myocardial ischemia is advanced to the stage of dilated poor LV because the symptoms are usually considered to be attributed to acute myocarditis/dilated cardiomyopathy1,2 or heart failure due to large shunts. Once the timely diagnosis is made, pushing the patient for surgical option and more so the execution of coronary surgery at such a delicate age in our set up is much more than a gigantic task.
CASE REPORT
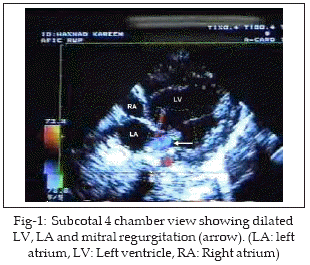
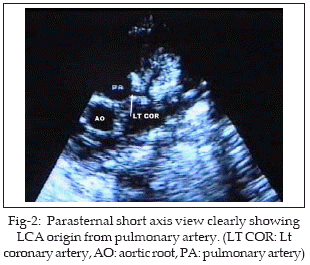
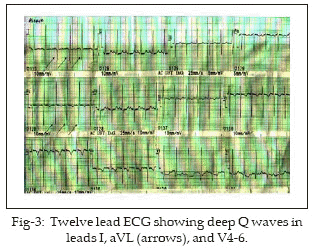
HK 2˝ months old baby boy was referred to our unit for echocardiography. There was history of increasing dyspnoea, feeding difficulty and irritability of three weeks duration. The diagnosis by referring physician was acute myocarditis with heart failure and poor response to inotropes, diuretics and antibiotics. Physical examination showed 4kg pink baby with tachycardia (HR 146/min), tachypnoea (RR 54/min), alae flaring and subcostal retractions. Heart sounds were normal with no murmur. Chest X-ray showed cardiomegaly. Echocardiography showed poor LV function with ejection fraction of 35%. Left sided chambers were dilated with mild mitral regurgitation (MR) (Fig-1). Parasternal short axis view clearly demonstrated the left coronary artery arising from pulmonary trunk and dividing into anterior descending and circumflex arteries (Fig-2). Twelve lead ECG showed evidence of anterolateral myocardial infarction with deep Q waves in I, aVL and V4-6 (Fig-3). A diagnosis of ALCAPA was made noninvasively and baby put on medications (aspirin, digoxin and furosamide-spironolactone combination). He was discussed with cardiac surgeon for early surgery but the parents did not agree. The baby is now 4˝ months old, coming regularly for follow up and has some symptomatic improvement.
DISCUSSION
Left coronary artery arising from pulmonary trunk is a rare but potentially lethal congenital heart disease with estimated incidence of 1/300,000 live births.2,3 Bland et al.1-3 in 1933 were first to correlate its clinical and autopsy findings on an infant. Thus the anomaly is also called Bland-White-Garland syndrome. In the fetus this condition is silent because blood supply to the LV myocardium is adequate due to high pulmonary artery pressures (PAP) and normal antegrade flow in the LCA.1 After birth the pulmonary artery contains desaturated blood but as long as the high PAP ensure the antegrade flow from PA to LCA, the baby remains asymptomatic.1-3 With postnatal drop in the PAP the antegrade flow into LCA decreases and eventual flow reversal into the PA occurs resulting in a "coronary steal".2 Initially the myocardial vessels dilate to increase the flow but soon the coronary vascular reserve is exhausted and myocardial ischemia and infarction occurs. Collateral vessels between the right and left coronary arteries develop in attempt to perfuse the LV myocardium but their efficiency is diminished due to "coronary steal".4 This usually occurs after the first month of life. Of all the children born with ALCAPA, about 87% present in infancy and about 65-85% die before first birthday, usually after two months of age.1 About 10-15% remains symptoms free and reach adult life, most probably due to large dominant right coronary and well-developed collaterals, but even they are at high risk of sudden death at a mean age of 35 years.2 The oldest reported case with the disease is a 72 years old woman.5 Typically the baby is well till 4-6 weeks of age and then gradually presents with profuse sweating, dyspnoea, pallor and irritability on feeding or crying (angina equilant),1,2,6 as was the presentation of our case. Physical examination may reveal signs of heart failure and/or mitral regurgitant murmur at the apex or even normal cardiac findings.1,2 Clinical diagnosis is extremely difficult even for the experienced physician because many of the more common conditions like acute myocarditis, dilated cardiomyopathy and even large L to R shunts & mixing lesion with unrestricted pulmonary flow may have similar presentation at this age. Standard investigation for the diagnosis is aortic root angiogram2,6 but solely for diagnostic purpose, that remains quite risky on an unstable patient. Therefore many people are of the view that ALCAPA should be diagnosed by noninvasive tools.2 On ECG, broad and deep Q waves in lead I and aVL with absent Q in II, III and aVF is considered characteristic of ALCAPA.7 On 2-D echocardiography the abnormal origin of LCA can be visualized.2 Colour flow Doppler can detect an abnormal jet of LCA into PA. A dilated right coronary artery (if present) may also be helpful in the diagnosis.2 Combining ECG and echocardiography ALCAPA can be diagnosed with reasonable certainty and with no need of angiography.8 In our case we clearly demonstrated on the echocardiography the abnormal origin of LCA from pulmonary trunk and the classic Q waves on ECG. Treatment options include LCA reimplantation and Takeuchi’s transpulmonary baffle; both types establish dual coronary systems.2 Sometimes on a very sick unstable patient simple ligation of the LCA at origin is adequate to stop the "coronary steal" and stabilize the patient for later more optimal surgery.9 Surgery should be performed even if the LV function is poor because LV function improves and reaches normal within a year with restoration of dual coronary supply.10,11
We conclude that there should be a high index of suspicion for ALCAPA during workup of any infant or child with dilated poor LV. Diagnosis by noninvasive tools like echocardiography in collaboration with classic ECG changes is reasonably straightforward. Surgical option should be offered to every patient irrespective of the patient’s age or LV condition.
REFERENCES
1. Matherne GP. Congenital anomalies of the coronary vessels and the aortic root. In: Allen HD, Clark EB, Gutgesell HP and Driscoll DJ (ed). Moss and Adam’s Heart disease in infants, children and adolescents 6th edition. Philadelphia, Lippincott William &Wilkins, 2001; 675-88.
2. Dodge-Khatami A, Mavroudis C, Backer CL. Anomalous origin of left coronary artery from pulmonary artery: Collective review of surgical therapy. Ann Thorac Surg 2002; 74: 946-55.
3. Mavroudis C, Dodge-Khatami A, Backer CL. Coronary artery anomalies. In: Mavroudis C and Backer CL (edi). Pediatric Cardiac Surgery 3rd edition. Philadelphia, Mosby 2003; 660-88.
4. Di Salvo G, Eyskens B, Claus P, D’hooge J, Bijnens B, Suys B, et al. Late post-repair ventricular function in patients with origin of left main coronary artery from the pulmonary trunk. Am J Cardiol 2004; 93: 506-8.
5. Fierens C, Budts W, Denef B, Van de Werf F. A 72 years old woman with ALCAPA. Hearts 2000; 83: 2.
6. Chien KJ, Chang WW, Lee CL, Huang TC, Lin CC, Hsieh KS, et al. Anomalous origin of the left coronary artery from the pulmonary artery-two cases report. Acta Cardiol Sin 2003; 19: 195-200.
7. Johnsrude CL, Perry JC, Cecchin F, Smith EO, Fraley K, Friedman RA, et al. Differentiating anomalous left main coronary artery originating from the pulmonary artery in infants from myocarditis and dilated cardiomyopathy by electrocardiogram. Am J Cardiol 1995; 75: 71-4.
8. Chang PR, Allada V. Electrocardiographic and echocardiographic features that distinguish anomalous origin of left coronary artery from pulmonary artery from idiopathic dilated cardiomyopathy Pediatr Cardiol 2001; 22: 3-10.
9. Kreutzer C, Schlichter AJ, Roman MI, Kreutzer GO. Emergency ligation of anomalous left coronary artery from the pulmonary artery. Ann Thorac Surg 2000; 69: 1591-2.
10. Schwartz ML, Jonas RA, Colan SD. Anomalous origin of left coronary artery from the pulmonary artery: recovery of left ventricular function after dual coronary repair. J Am Coll Cardiol 1997; 30: 547-53.
11. Pandey R, Ciotti G, Pozzi M. Anomalous origin of left coronary artery from pulmonary artery: result of surgical correction in five infants. Ann Thorac Surg 2002; 74: 1625-30.
HOME | SEARCH | CURRENT ISSUE | PAST ISSUES
Professional
Medical Publications
Room No. 522, 5th Floor, Panorama Centre
Building No. 2, P.O. Box 8766, Saddar, Karachi - Pakistan.
Phones : 5688791, 5689285 Fax : 5689860
pjms@pjms.com.pk