|
|
||||
|
Published by : PROFESSIONAL MEDICAL PUBLICATIONS |
||||
|
ISSN 1681-715X |
||||
|
||||
|
- |
||||
|
CASE REPORT |
||||
|
- |
||||
|
Volume 23 |
July - September 2007 |
Number 4 |
||
|
|
||||
|
||||
|
|
||||
|
Published by : PROFESSIONAL MEDICAL PUBLICATIONS |
||||
|
ISSN 1681-715X |
||||
|
||||
|
- |
||||
|
CASE REPORT |
||||
|
- |
||||
|
Volume 23 |
July - September 2007 |
Number 4 |
||
|
|
||||
|
||||
Alpers disease: Report of two familial cases
M. Barzegar1, Mazyar Hashemilar2
ABSTRACT
Alpers syndrome is usually characterized by a clinical triad of psychomotor retardation, intractable epilepsy and liver failure in infants and young children. It is a hereditary disease with an autosomal recessive pattern of inheritance. Definitive diagnosis is shown by postmortem examination of the brain and liver. There is no known treatment. In this article two familial cases are reported.
KEY WORDS: Alpers syndrome, Neuronal degeneration, Liver disease, Progressive cerebral poliodystrophy.
Pak J Med Sci July - September 2007 Vol. 23 No. 4 643-646
1. M. Barzegar,
Associate Professor of Pediatric Neurology,
Tabriz Children’s Hospital,
2. Mazyar Hashemilar,
Assistant Professor of Neurology,
1-2: Tabriz University of Medical Sciences,
Tabriz – Iran.
Correspondence
Mazyar Hashemilar,
Razi Hospital,
El Goli Road,
Tabriz – Iran.
E-mail: mhashemilar@yahoo.com
* Received for Publication: March 19, 2007
* Revision Received: March 24, 2007
* Accepted: June 5, 2007
INTRODUCTION
Alpers disease, a syndrome of unknown etiology, represents a group of disorders characterized by a rapidly progressive encephalopathy with intractable seizures and diffuses neuronal degeneration. In most of patients liver involvement is present and an autosomal recessive pattern of transmission is suggested. A diagnosis of Alpers disease should be made only in the absence of known metabolic disease or an antecedent event. Along with clinical features suggestive of Alpers disease, neuroimaging should exclude other diagnostic possibilities and should show progressive brain atrophy on successive studies, with relative sparing of the white matter. Seizure types include myoclonic, focal, and generalized tonic clonic convulsions. Treatment is supportive and prognosis is poor.
CASE REPORT
An eighteen months old boy was admitted to our centre due to progressive psychomotor deterioration, hypotonia and seizures since seven months ago. He has been healthy up to eleven months old, with normal developmental milestones and beginning to walk with help. His mother and father had a far familial relationship and did not have any neurologic problem. They have two healthy children, a fifteen years old daughter and a three years old son. Their second child, a girl had died when fourteen moths old, following a three months period of illness. Her disease had begun with seizures. A refractory status epilepticus had occurred during the course of her disease which was controlled by sodium valproate. Afterwards she had become flaccid, had repeated vomiting bouts and become jaundiced. Valproic acid has been stopped following results of liver enzyme assays which showed significant elevation. Her progressive mental deterioration, followed by generalized myoclonic status and flaccidity. She had died following aspiration pneumonia three months after onset of illness. A brain CT scanning had shown cerebral atrophy.
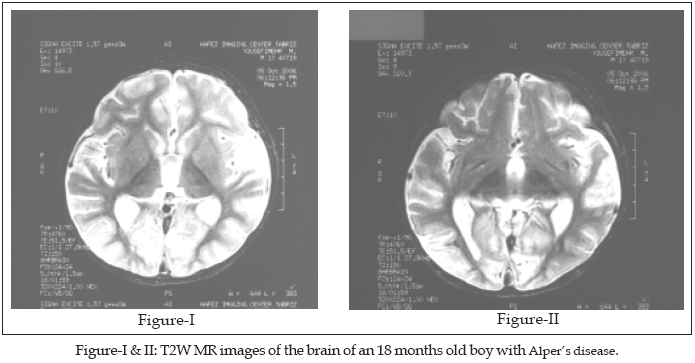
Our patient’s illness had begun seven months ago with sudden onset of focal myoclonic jerks which was culminated in a generalized myoclonic status epilepticus three days later and hospitalization. A brain CT scan had shown no abnormality at that time. Afterwards he became hypotonic, so he could not sit without support and even lost his head control. His seizures were refractory to treatment with phenobarbital, vigabatrin, primidone, clonazepam, nitrazepam, carbamazepine, vitamin B6, phenytoin and a course of ketogenic diet. Gradually he became less alert and attentive and had difficult swallowing. He was admitted to hospital due to aspiration pneumonia afterwards. One month ago he developed repeated vomiting bouts without any apparent cause. During his current admission, his seizures were controlled with midazolam infusion, phenobarbital and phenytoin in full dose. He was hypotonic, deep tendon reflexes were responsive, plantar responses abolished. His eye movements showed no limitation, but following of visual objects were impaired apparently. Fundoscopic examination showed no abnormality and direct and indirect pupillary light responses were normal. Comprehensive biochemical work-up, including: serum lactate, pyruvate, ammonia, copper, magnesium, ceruloplasmin, phenylalanine, thyroid function tests, 24 hour urine copper excretion and urine reducing substances showed no abnormality. A complete blood count and blood coagulation profile was normal. The liver enzymes showed moderate elevation. The microscopic examination of the patient’s hair showed no abnormality in favor of Menkes disease. The electromyography and nerve conduction studies showed no evidence of lower motor neuron involvement. The electroencephalogram showed a diffusely slow (4-5 Hz) background, superimposed on which were random spikes and sharp wave activity. The brain MR imaging was remarkable for diffuse cerebral atrophy, considerable decrease in white matter and cortical thinning of the frontal, posterior temporal and occipital lobes. Occipital lobe atrophy is severe and there are bilateral symmetrical T2W hypointense signal abnormalities in both thalami. (Figures I and II).
DISCUSSION
Alper’s disease usually begins in early life with convulsions. A progressive neurologic disorder characterized by spasticity, myoclonus, and dementia ensues. Status epilepticus is often the terminating development. Bernard Alpers in 1931 described the neuropathology and clinical features in a 4-month-old girl with a one-month illness characterized by intractable generalized seizures. He termed the disorder ‘diffuse progressive degeneration of the gray matter of the cerebrum’.
1 Huttenlocher et al.2 Noted that hepatic involvement was absent in some cases reported earlier, including the case reported by Alpers.1 Harding reviewed the clinical, neurologic, electrophysiologic, and histopathologic features of Alpers syndrome in 32 patients.3 Birth was usually normal, with some developmental delay in infancy, often with hypotonia and bouts of vomiting. The seizure disorder usually had an abrupt onset and although clinical signs of liver disease often appeared later, biochemical evidence of liver disease was sometimes present before the onset of seizures. EEG and visual evoked potentials were abnormal. Most patients died before the age of three years. Less frequently, late presentation occurred, even up to 25 years of age. Some patients also had visual disturbances. Liver pathologic findings, including fatty changes, abnormal bile duct architecture, and fibrosis, were unrelated to anticonvulsant therapy. Neuropathology showed severe cortical neurodegeneration and astrocytosis. In 12 of the 26 families in their series, two or three siblings were affected, including one pair of twins. Frydman et al. reported the cases of eight patients from two families.4 Onset in the first family was prenatal; in the four patients who were examined, severe microcephaly, intrauterine growth retardation and typical manifestations of fetal akinesia, including retrognathia, joint limitations and chest deformity, were found. The second family presented with an early infantile form. All of the affected offspring had micrognathia and one had findings of fetal akinesia, comparable to those seen in the other family. Microcephaly was mild at birth and progressed with age. Refractory neonatal convulsions, swallowing difficulties, and pneumonia complicated the clinical course of patients in both families and all of the infants died before age 20 months. Comprehensive biochemical and metabolic studies in both families yielded normal results, and the diagnosis was supported by demonstration of extensive progressive brain atrophy on computerized tomography and typical histologic findings; for example, the parietal cortex showed spongy state with focally accentuated severe loss of neurons. The cerebellar cortex showed severe loss of almost all granular cells and persistent Purkinje cells. Anomalies of dendritic arborization were also seen. Both families were of Israeli Arab ethnicity and the parents were first cousins in both cases. Harding et al.3 reported the unusual cases of two unrelated girls, aged 17 and 18, with a progressive encephalopathy, visual signs and symptoms, multiple types of drug-resistant seizures and liver failure. Brain imaging showed lesions in the occipital lobe, and EEG showed slow waves with polyspikes. Both patients had a rapid degenerative course and died within 8 months of onset.The mode of transmission of Alpers disease is understood and it is consistent between families. Case studies and biochemical evidence support autosomal recessive inheritance and maternal or mitochondrial inheritance patterns. Cases with autosomal-recessive inheritance patterns have no mitochondrial deficiencies.
3Recently compound heterozygosity for 2 mutations in the POLG gene is reported in several cases of Alpers disease.
4-6 Liver biopsies from some of these patients has shown mitochondrial DNA depletion ranging from 87 to 94% and decreased activity of mtDNA-encoded respiratory chain complexes.6The electroencephalographic findings show a pattern of continuous, anterior, high-voltage, l-to-3-Hz spike-and-wave like activity that persists despite intermittent focal seizures. Also there is a progressive slowing of background activity as the disease progresses.
7 An epilepsia partialis continua and convulsive status epilepticus is also seen in these patients.8Cranial CT scans demonstrate progressive atrophy and low densities in occipital and temporal lobes. There is involvement of cortex and white matter. Generalized atrophy is common throughout the brain in the later stages of the disease. Multiple findings are apparent on conventional MRI scans. These include diminished white matter and cortical thinning of the frontal posterotemporal and occipital lobes.
9 Lesions of the thalamus also have been reported.10The cases reported here in two siblings of different sexes show an autosomal pattern of inheritance. The MR imaging findings are remarkable for the severity of the involvement of the occipital lobes and the thalami.
REFERENCES
1. Alpers BJ. Diffuse progressive degeneration of gray matter of cerebrum. Arch Neurol Psychiat 1931;25:469-505.
2. Huttenlocher PR, Solitare GB, Adams G. Infantile diffuse cerebral degeneration with hepatic cirrhosis. Arch Neurol 1976;33(3):186-92.
3. Harding BN. Progressive neuronal degeneration of childhood with liver disease (Alpers-Huttenlocher syndrome): A personal review. J Child Neurol 1990;5(4):273-87.
4. Naviaux RK, Nguyen KV. POLG mutations associated with Alpers syndrome and mitochondrial DNA depletion. (Letter) Ann Neurol 2005;58(3):491.
5. Davidzon G, Mancuso M, Ferraris S, Quinzii C, Hirano M, Peters HL, et al. PLOG mutations and Alpers syndrome. Ann Neurol 2005;57(6):921-4.
6. Ferrari G, Lamantea E, Donati A, Filosto M, Briem E, Carrara F, et al. Infantile hepatocerebral syndromes associated with mutations in the mitochondrial DNA polymerase-gamma A. Brain 2005;128(4):723-31.
7. Frydman M, Jager-Roman E de Vries L, Stoltenburg-Didinger G, Nussinovitch M, Sirota L. Alpers progressive infantile neuronal poliodystrophy: An acute neonatal form with findings of the fetal akinesia syndrome. Am J Med Genet 1993;47(1):31-6.
8. Brick IE, Westmoreland Ill, Gomez MR. The electroencephalogram in Alper’s disease [abstract]. Electroencephalogr Clin Neurophysiol 1984;58:31.
9. Walton A. A case study of Alper’s disease in siblings. Am J EEG Technol 1996;36:18-27.
10. Barkovich AJ, Good WV, Koch TK, Berg BO. Mitochodrial Disorders: Analysis of their clinical and imaging characteristics. Am J Neuroradiol 1993;14(5):1119-37.
HOME | SEARCH | CURRENT ISSUE | PAST ISSUES
Professional
Medical Publications
Room No. 522, 5th Floor, Panorama Centre
Building No. 2, P.O. Box 8766, Saddar, Karachi - Pakistan.
Phones : 5688791, 5689285 Fax : 5689860
pjms@pjms.com.pk